Diffusion-weighted imaging (DWI)
Last updated on 2024-09-24 | Edit this page
Overview
Questions
- What does diffusion weighted imaging measure?
- What processing steps are needed when working with diffusion weigthed imaging data?
- What types of analyses are used with diffusion imaging in dementia resaerch?
Objectives
- Understand the processing steps in diffusion-weighted MRI scans
- Perform basic analyses on white matter microstructure.
Introduction
We will use the FSL diffusion toolbox to perform the processing steps that are core to most diffusion weighted imaging analyses:
- image visualization of raw data and analysis outputs,
- eddy correction,
- generation of diffusion tensor metrics brain maps,
- tract-based spatial statistics,
- you can also stretch your knowledge and familiarize yourself with loops to run these commands on multiple subjects, all shown at the end of this tutorial
We are going to be working in the DiffusionMRI subfolder under data
in your home directory, ~/data/DiffusionMRI.
Looking at the raw data
From the previous lessons, you learned how to view and navigate
images, let’s first look at the raw data, which can all be found under
~/data/DiffusionMRI/sourcedata.
To go to this directory using the terminal, use the command
cd to change directory. Type
cd ~/data/DiffusionMRI/sourcedata to go this
directory.
Let’s inspect what each participant’s dwi directory should contain:
or
The * will match any following text, which here will
list the contents of any directory starting with “sub-OAS”.
Each directory should contain 4 files:
- one
.bvaltext file - one
.bvectext file - one
.jsontext file - one nifti (
.nii.gz) image file
All files are required for processing DWI data except the .json file.
The .json file is specific to the BIDS data organization, and is
a useful way to access data description. More and more software are also
relying on data organized according to the BIDS structure.
Let’s look at those files:
BASH
# Change directory to go in one participant's folder
cd sub-OAS30001/dwi
## Image file
fsleyes sub-OAS30001_dwi.nii.gz
## Text files
cat sub-OAS30001_ses-d2430_dwi.bval
cat sub-OAS30001_ses-d2430_dwi.bvec
- The nifti file is a 4D file of all the directions acquired.
- The
.bvalfile refers to the b-value applied to each image. - The
.bvecfile refers to the vector applied to each image, with the coordinates in x, y and z.
Refer to the Getting started session for more details!
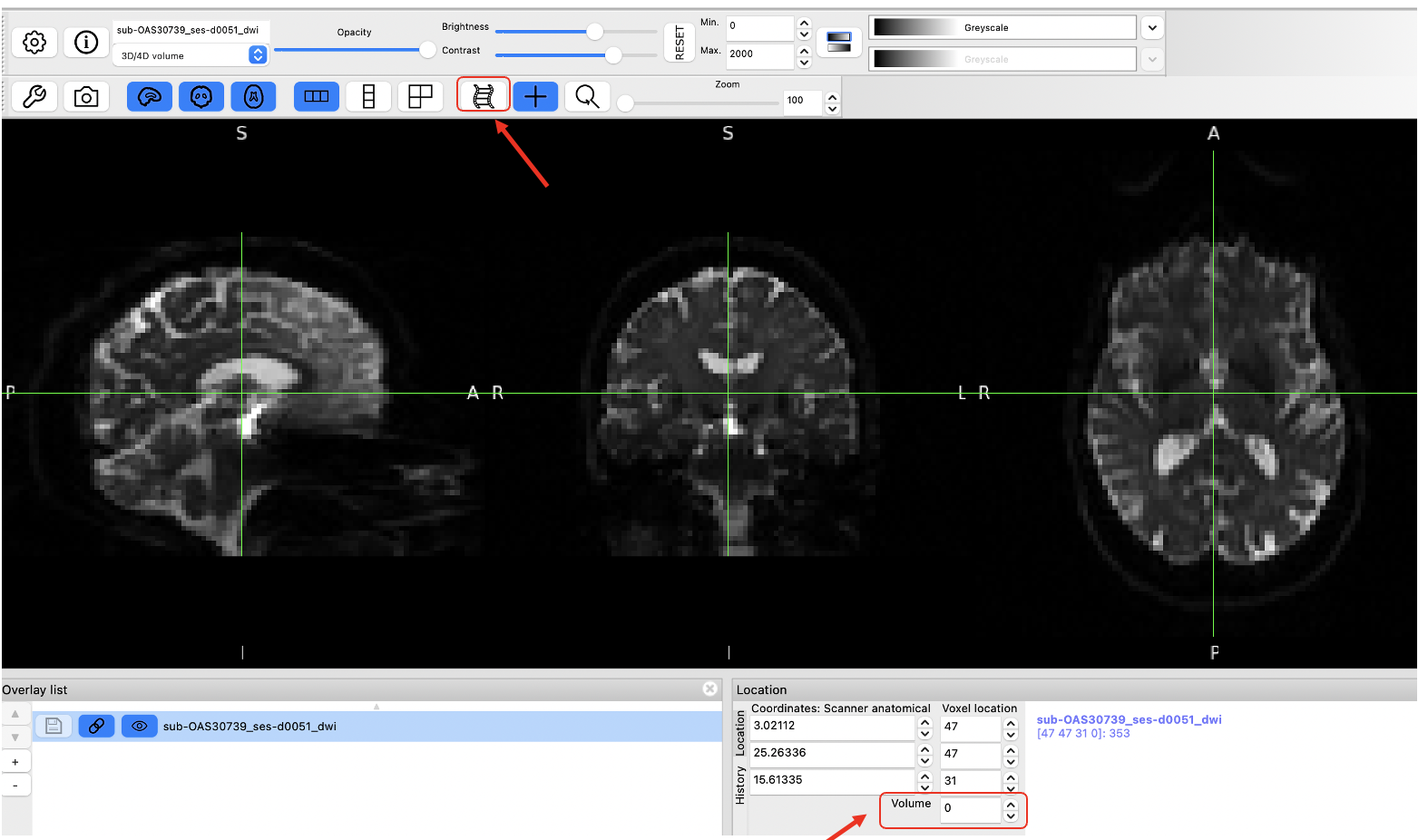
There is 1 b0 image and 64 gradient images! You can check for the maximum number as you move through the “Volume” box as shown below.
Data preprocessing
We are now ready to start data pre-processing, which we will do using the FSL Diffusion Toolbox. Most of the steps and explanation below are adapted from the excellent tutorial provided FSL, please refer to it for more details: https://open.win.ox.ac.uk/pages/fslcourse/practicals/fdt1/index.html
All the steps below will be done on one subject only, but if you wish to loop the different steps across multiple participants, refer to the section Stretch your knowledge at the end.
1. Creating a brain mask from the b0 images
We often use the b0 images (there is only 1 b0 image in this dataset) to create a brain mask that is needed in future steps. First select the b0 image, extract the brain only and binarize it to make a mask.
-
Extract the b0 image only with select_dwi_vols or fslroi
-
Brain extraction and binarization with bet
-
Load the mask you just created to make sure it is adequate!
2. Correcting for susceptibility-induced distortions
Some parts of the brain can appear distorted depending on their magnetic properties. One common way to correct the distortions with DWI data is by acquiring a b0 image acquired with a different phase-encoding, and merging the two types of images running TOPUP.
In this dataset we don’t have the data required for TOPUP so we will skip this step.
Note however that you should run it if your data allows.
3. Correcting for eddy currents and movement
Eddy is a tool to correct for eddy current-induced distortions and movement on the image. Eddy currents arise from electric current due to strong and fast changing gradients. Eddy also does outlier detection and will replace signal loss by non-parametric predictions.
We need to create 2 files to be able to run eddy:
- an index file corresponding to the directions with the same phase encoding
- a file with some acquisition parameters
Run the following lines to create the index file. Since all images are obtained with the same phase encoding, it will just be a vector with values of 1 of the same length of the bval file.
BASH
## The command wc (wordcount) will check the length of the bval file and we will use this output to create the index file we need.
wc -w sub-OAS30001_dwi.bval
indx=""
for ((i=1; i<=65; i+=1)); do indx="$indx 1"; done
echo $indx > index.txtOUTPUT
1 1 1 1 1 1 ... 1
The index file is a series of 1 repeated 65 timesThe acquisition parameter file is a vector containing the phase
encoding and the total read out time, which can all be found in the
.json file. Do cat sub-OAS30001_dwi.json and
see if you can find the following information.
- TotalReadoutTime: 0.0451246
- PhaseEncodingDirection: j- : This corresponds to the AP direction and is coded as -1 for the acquisition parameter file.
You can also try
cat sub-OAS30001_dwi.json | grep 'Total' to directly find
the entry you need!
The acquisition parameter file first includes the phase encoding, with the first 3 numbers corresponding to the x, y, and z directions. Here we are acquiring along the y direction (being -1), and x and z being 0. The last number is the total read out time.
To create it you can do:
We are ready to run eddy! Please refer to https://fsl.fmrib.ox.ac.uk/fsl/fslwiki/eddy for all the details
However, eddy takes a long time to run (about 40 minutes either on
the VM or on a MacBook Pro), so we’ve run it for a few subjects already.
The outputs can be found in
~/data/DiffusionMRI/processed_sourcedata.
As eddy creates a lot of output files, it can be good practice to
create a separate directory to store the outputs so we keep things more
organized, as done in the processed_sourcedata directory.
For example, if you type
ls ~/data/DiffusionMRI/processed_sourcedata/sub-OAS30001/eddy/
you will see all the eddy outputs for this given participant.
Eddy also takes a lot of input arguments, as depicted below
Image adapted from https://open.win.ox.ac.uk/pages/fslcourse/practicals/fdt1/index.html
If you want to try to run it on one participant, you can try the following command.
BASH
eddy --imain=sub-OAS30001_dwi.nii.gz --mask=sub-OAS30001_b0_bet_mask.nii.gz --acqp=acqparams.txt --index=index.txt --bvecs=sub-OAS30001_dwi.bvec --bvals=sub-OAS30001_dwi.bval --out=../eddy/eddy_correctedTo be able to run the next step, we will copy the eddy-corrected DWI
scan for one subject into our working directory. Make sure you are in
this directory:
~/data/DiffusionMRI/sourcedata/sub-OAS30001/dwi (you can
use the command pwd to print your working directory and
know where you are), and then type this command:
BASH
cp ~/data/DiffusionMRI/processed_sourcedata/sub-OAS30001/eddy/sub-OAS30001_eddy_corrected.nii.gz .Now let’s compare the raw DWI scan vs. after eddy correction. You can
load the two images (sub-OAS30001_dwi.nii.gz and
sub-OAS30001_eddy_corrected.nii.gz) with fsleyes. On this
participant with high quality images, the differences are not really
noticeable, but remember the example from the webinar on data where
there was signal loss and big differences were evident.
** It’s always important to inspect images after preprocessing steps! Many software (including FSL) have automated QC frameworks available to help go through all the outputs. You can find more information on eddyQC from the FSL documentation if you want to try it
4. Generating DTI outputs
We can now fit the diffusion tensor model to the preprocessed data. This will generate all the standard DTI outputs, like fractional anisotropy (FA), mean diffusivity (MD), radial diffusivity (RD) and axial diffusivity (AD), along with eigenvectors.
As this will also generate many outputs, to keep things organized let’s create a directory for storing the outputs.
The command is dtifit, and uses the eddy-corrected data
as input. All the inputs required are detailed in the command usage.
BASH
#Usage: Compulsory arguments (You MUST set one or more of):
# -k,--data dti data file
# -o,--out Output basename
# -m,--mask Bet binary mask file
# -r,--bvecs b vectors file
# -b,--bvals b values file
dtifit --data=sub-OAS30001_eddy_corrected.nii.gz --mask=sub-OAS30001_b0_bet_mask.nii.gz --bvecs=sub-OAS30001_dwi.bvec --bvals=sub-OAS30001_dwi.bval --out=../dti/sub-OAS30001_Many files have been created, let’s look at the ones most commonly
used, i.e. FA, MD and V1.
V1 is the principal eigenvector and corresponds to the direction along
the principal diffusion direction and allows us to visualize the
underlying orientation of white matter fibers.
V1 should open as an RGB map where the colors represent
directions:
* Red= left - right axis
* Green= anterior - posterior axis
* Blue= superior - inferior axis
You can also change the overlay type and visualize the image as lines with ‘3-direction vector image (Line)’ in the top left corner, which will show the vector field.
Example of the V1 file in RGB:
We have now completed the basic steps required for all diffusion data! Those allow to continue with further processing (e.g. tractography) or to continue with group-level analyses, which require processing a few subjects.
In the next section, you have a few options to go further:
- There is some example of code to perform the same steps as we did above, this time looping across subjects.
- A common analyses is Tract-Based Spatial
Statistics (TBSS), which allows for voxel-wise analyses on a
skeletonized white matter template. We provided the command lines below
to do such analyses. Some steps can take some time to run, and thus we
provided the outputs in
~/data/DiffusionMRI/tbssif you want to examine the different steps and the final outputs.
Stretch your knowledge
Do you want to process multiple subjects?
An easy way to do this is through multiple bash loops, introduced this morning. You will have examples below of loops for the different steps we did above if you want to try it.
Make sure you are in the following directory:
~/data/DiffusionMRI/sourcedata
BASH
# Extract the b0 image
for x in sub-OAS30*; do echo $x; fslroi $x/dwi/*_dwi.nii.gz $x/dwi/${x}_b0.nii.gz 0 1; done
#Create the brain mask from the b0 image
for x in sub-OAS30*; do echo $x; bet $x/dwi/*_b0.nii.gz $x/dwi/${x}_b0_bet -m; done
# Run eddy - Don't try this on multiple subjects; it will take too long to run!
for x in sub-OAS30*; do echo $x; eddy --imain=$x/dwi/${x}_dwi.nii.gz --mask=$x/dwi/${x}_b0_bet_mask.nii.gz --acqp=acqparams.txt --index=index.txt --bvecs=$x/dwi/${x}_dwi.bvec --bvals=$x/dwi/${x}_dwi.bval --out=$x/dwi/${x}_eddy_corrected; done
# Run dtifit
for x in sub-OAS30*; do echo $x; dtifit --data=$x/dwi/${x}_eddy_corrected.nii.gz --mask=$x/dwi/${x}_b0_bet_mask.nii.gz --bvecs=$x/dwi/${x}_dwi.bvec --bvals=$x/dwi/${x}_dwi.bval --out=$x/dwi/${x} ; doneNote that all processed data can be found in
~/data/DiffusionMRI/processed_sourcedata if you want to
access it
Do you want to investigate microstructure differences between groups?
Let’s investigate FA differences between 4 Controls and 4 AD subjects using TBSS. This involves a few easy steps, which are outline below, but please refer to the original description of TBSS for all details.
It can take some time to run, so we placed all outputs in
~/data/DiffusionMRI/tbss if you want to explore the outputs
from each step.
If you want to try it on your own, you can follow these steps:
-
Create a new directory for TBSS and copy all FA maps in it
BASH
mkdir tbss_tutorial cp ~/data/DiffusionMRI/processed_sourcedata/sub-OAS30*/dti/*FA.nii.gz tbss_tutorial/You can run
slicesdir *.nii.gzand open the resulting web page report; this is a quick way to check your images.Run all the next commands directly from the tbss_tutorial directory you created.
-
Quick preprocessing of the data
-
Register all FA maps to standard space
-
Apply transformation to all images and create mean FA skeleton
You can load the 4D file created
all_FAwith the overlaymean_FA_skeletonon top to verify that all images are aligned correctly. -
threshold FA skeleton
This last step is to threshold the FA skeleton that was created, inspect it before and adapt accordingly (here we used the recommended 0.2 threshold)
We are now ready to do the statistical comparisons!
Statistical comparisons between two groups
We will use the tool randomise, for which we need to create two text files: design.mat and design.con. Design.mat specifies which groups the participants belong to and other covariates of interest. Design.con specifies the contrasts we want to perform for our statistical analyses.
You need to make sure of the order of your FA images to create the
design matrix files, which is the alphabetical order. In this example,
we will compare Controls vs. AD patients. The diagnosis for each
participant is as follows:
sub-OAS30001 : Control
sub-OAS30003 : Control
sub-OAS30006 : Control
sub-OAS30024 : AD
sub-OAS30025 : Control
sub-OAS30128 : AD
sub-OAS30217 : AD
sub-OAS30262 : AD
We want to perform 2-sample t-test. We can create the files we need easily with:
Since the order of our participants do not perfectly follow
the Diagnosis categories, make sure you fix the design.mat accordingly
if you used the automated command. The final one should
be:
1 0
1 0
1 0
0 1
1 0
0 1
0 1
0 1
For the design.con, the first contrast ‘1 -1’ will give results where CU > AD and the second contrast ‘-1 1’ will give results where the CU < AD. Remember: we are looking at differences in FA, so we expect smaller FA values in AD than Controls. It would be the opposite if we were looking at differences in MD.
We are ready do run the command randomise to compare
groups. There are a lot of options you can choose to generate your
results, with different thresholding options. Please refer to the full
description of the command
BASH
#Usage: randomise -i <input> -o <output> -d <design.mat> -t <design.con> [options]
# Since we have only a few subjects we can apply a less stringent threshold, setting a cluster-based threshold at t= 1.5
randomise -i all_FA_skeletonised -o tbss -m mean_FA_skeleton_mask -d design.mat -t design.con -c 1.5
# Here is the example to apply the preferred method of TFCE (threshold free cluster enhancement; option T2)
randomise -i all_FA_skeletonised -o tbss -m mean_FA_skeleton_mask -d design.mat -t design.con -n 100 --T2 -VWe generated outputs using the two methods (cluster based threshold
of TFCE), which you can access under
~/data/DiffusionMRI/tbss/stats. You have:
* unthresholded t-statistical maps,
tbss_tstat1 and tbss_tstat2, where 1 and 2 refer to the
different contrasts
* p-values maps corrected for multiple comparisons (either
tbss_clustere_corrp_tstatX or tbss_tfce_corrp_tstatX)
Note that the p-values maps are outputted as 1-p for convenience of
display (so that higher values are more significant). You can threshold
those from 0.95 to 1.
Here is one example of how you can overlay different outputs images to visualize results.
BASH
fsleyes -std1mm mean_FA_skeleton -cm green -dr .3 .7 tbss_tstat1 -cm red-yellow -dr 1.5 3 tbss_clustere_corrp_tstat1 -cm blue-lightblue -dr 0.90 1This will display the standard MNI brain, the average FA skeleton used for TBSS, the t-map for contrast 1 and the p-value maps after cluster-based thresholding. Note that I am thresholding the latter from 0.90 to 1 since we have weak results in this example; this will show voxels where CU have greater FA than AD patients from p=0.10.
Try the same thing with the other contrast to confirm if results are as expected.
Key Points
- Diffusion imaging is used to obtain measurements of tissue microstructure
- DWI consists of a 4D volume, and files describing the vectors and b-values that each volume is encoding
- The common core processing steps are: brain masking, susceptibility correction, eddy/motion correction, and modelling (tensor fitting, NODDI, etc)